
Síndromes mielodisplásicos en niños
Hallazgos recientes e implicaciones
Los síndromes mielodisplásicos (SMD) en niños han ganado cada vez más atención debido a los avances recientes en la comprensión de su etiología, clasificación y asociación con los síndromes hereditarios de insuficiencia de la médula ósea. Este artículo revisa los últimos hallazgos en MDS pediátricos, centrándose en la clasificación más reciente que abarca la citopenia refractaria de la infancia (CCR), MDS avanzado y MDS relacionados con la terapia. Además, explora los desafíos planteados por la frecuente co-ocurrencia de trastornos hematopoyéticos clonales con síndromes hereditarios de insuficiencia de la médula ósea, lo que complica tanto el diagnóstico como el tratamiento. Además, se discute la identificación emergente de síndromes de línea germinal relacionados con el desarrollo de SMD y leucemia mieloide aguda, como mutaciones en GATA2, ETV6, SRP72 y SAMD9 / SAMD9-L.
En uno de mis primeros viajes a Guatemala, al mencionar este diagnóstico en uno de los pacientes del Hospital Roosevelt, uno de los más brillantes Pediatras que he conocido ahí, me buscó para preguntarme si estaba seguro de lo que estaba mencionando. Y es que la costumbre era la de tener reservado estos diagnósticos exclusivamente para adultos y era algo que estaban escuchando por primera vez. Comprendí su posición y su situación, pero no podía hacer nada por traer a todo el equipo a esta nueva etapa de la Medicina y de la Especialidad de la Hematología Pediátrica.
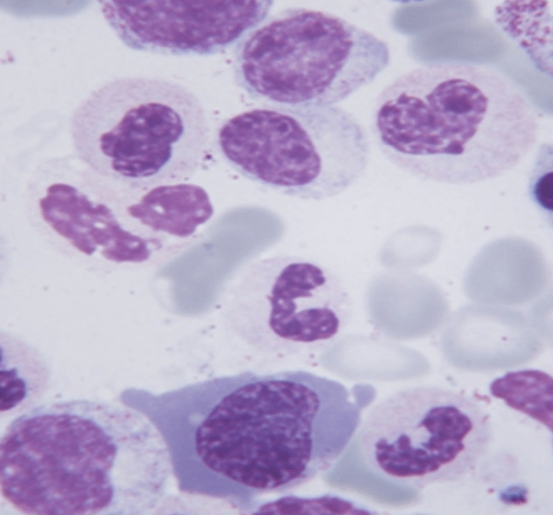
SMD
Los síndromes mielodisplásicos (SMD) son un grupo heterogéneo de trastornos hematopoyéticos clonales caracterizados por hematopoyesis displásica e ineficaz.
Tradicionalmente consideradas enfermedades de los adultos, los SMD también han sido reconocidos en la población pediátrica.Investigaciones recientes han ampliado nuestra comprensión de los SMD pediátricos, arrojando luz sobre nuevos criterios de clasificación, asociaciones con síndromes hereditarios de insuficiencia de la médula ósea y la identificación de síndromes de la línea germinal que predisponen a los SMD y la leucemia mieloide aguda.
Clasificación reciente de los SMD pediátricos
La clasificación más reciente de SMD pediátrico incluye tres subtipos:
La clasificación más reciente de SMD pediátrico incluye tres subtipos:
- 1. Citopenia infantil refractaria (CCR),
- 2. SMD avanzado y
- 3. SMD relacionados con el tratamiento.
1. El CCR se presenta con citopenia persistente y menos del 5% de blastocitos en la médula ósea. 2. El SMD avanzado se caracteriza por un aumento del recuento de blastocitos, que se asemeja a la leucemia mieloide aguda (LMA), mientras que el 3. SMD relacionado con el tratamiento surge como consecuencia de laquimioterapia previa o la exposición a la radiación.
Este refinado sistema de clasificación ayuda en el diagnóstico preciso y estrategias de gestión apropiadas.
Los niños con SMD con frecuencia exhiben una presencia concomitante de síndromes hereditarios de insuficiencia de la médula ósea (IBMFS), un grupo de trastornos genéticos caracterizados por hematopoyesis deteriorada. Esta co-ocurrencia plantea desafíos de diagnóstico, ya que las características clínicas superpuestas pueden confundir la identificación precisa.
Además, los casos de SMD asociados a IBMFS pueden presentarse con trayectorias de enfermedad distintas, lo que requiere enfoques de tratamiento personalizados y de Alta Precisión.
Los avances recientes en genética han revelado mutaciones de la línea germinal que predisponen a las personas a MDS y AML.
A. Las mutaciones en GATA2, un factor de transcripción crucial para la hematopoyesis, se han relacionado con SMD/LMA familiar, monocitopenia e inmunodeficiencia.
B. Las mutaciones en ETV6, asociadas con trombocitopenia familiar y neoplasias hematológicas, también están implicadas en el SMD pediátrico. Se han identificado mutaciones en SRP72 y SAMD9/SAMD9-L en casos sindrómicos de SMD, destacando la compleja interacción entre la genética y el desarrollo de la enfermedad.
Conclusión:
En mi humilde criterio, el panorama de los SMD pediátricos está evolucionando rápidamente, impulsado por los recientes descubrimientos en la clasificación, las asociaciones con los síndromes hereditarios de insuficiencia de la médula ósea y la identificación de síndromes de la línea germinal que aumentan la susceptibilidad a los SMD y la LMA. Estos hallazgos enfatizan la necesidad de colaboración multidisciplinaria para mejorar la precisión diagnóstica y desarrollar estrategias de tratamiento dirigidas. A medida que continúa la investigación, una comprensión más profunda de los fundamentos moleculares de los SMD pediátricos probablemente
conducirá a mejores resultados para los niños afectados.

Biografia
1. Síndrome mielodisplásico en niños y adolescentes.
Niemeyer CM, Baumann I. Semin Hematol. Ene. 2008;45(1):60-70. doi: 10.1053/j.seminhematol.2007.10.006. PMID: 18179970
2. Síndrome mielodisplásico infantil. Chatterjee T, Choudhry VP. Indian J
Pediatr. Sep. 2013:80(9):764-71. doi: 10.1007/s12098-013-1130-8. Epub 2013
3 de agosto. PMID: 23912822 Revisión
3. Síndromes de predisposición genética: ¿cuándo deben considerarse en la evaluación del SMD? Babushok DV, Bessler M. Best Pract Res Clin Haematol. Marzo de 2015;28(1):55-68. doi: 10.1016/j.beha.2014.11.004. Publicación electrónica, 12 de noviembre de 2014
Manejo de los síndromes mielodisplásicos pediátricos:
Enfoques y desafíos actuales
Los síndromes mielodisplásicos pediátricos (SMD) constituyen un grupo raro y complejo de trastornos clonales, que representan una pequeña fracción de las neoplasias hematológicas infantiles. Estos trastornos presentan desafíos únicos debido a su heterogeneidad y asociaciones con síndromes hereditarios de insuficiencia de la médula ósea. Además, los avances recientes han arrojado luz sobre los síndromes de la línea germinal que predisponen a las personas al SMD, como las mutaciones en genes como GATA2, ETV6, SRP72 y SAMD9 / SAMD9-L. En este artículo, profundizamos en las estrategias actuales para el manejo del SMD pediátrico, centrándonos en los enfoques de tratamiento y sus implicaciones.
Los SMD pediátricos son relativamente raros, con una incidencia anual de 1 a 4 casos por millón. Aunque comprenden menos del 5% de las neoplasias hematológicas infantiles, su complejidad requiere estrategias de manejo personalizadas. En particular, estos trastornos a menudo se manifiestan en el contexto de los síndromes hereditarios de insuficiencia de la médula ósea, presentando un aspecto distinto de los SMD en pacientes pediátricos.
Los avances recientes en genética han puesto de relieve el papel de las mutaciones de la línea germinal en la predisposición de los individuos a desarrollar SMD y leucemia mieloide aguda. Las mutaciones en GATA2, ETV6, SRP72 y SAMD9 / SAMD9-L han surgido como actores cruciales en la patogénesis del SMD pediátrico, arrojando luz sobre la intrincada relación entre la predisposición genética y el desarrollo de enfermedades.
Entre las variantes pediátricas de SMD, la citopenia refractaria de la infancia (CCR) se destaca como la más prevalente. Caracterizado por características histopatológicas específicas, el CCR plantea desafíos de tratamiento únicos. Para los pacientes con médula ósea hipocelular y la ausencia de monosomía 7 o un cariotipo complejo, se puede considerar la terapia inmunosupresora . Sin embargo, las tasas de respuesta son modestas en comparación con la anemia aplásica grave, y una proporción sustancial de pacientes puede requerir eventualmente un trasplante alogénico de células madre hematopoyéticas (TCMH) debido a la falta de respuesta o la recaída.
CONCLUSIÓN
El manejo de los síndromes mielodisplásicos pediátricos presenta un panorama clínico complejo, caracterizado por rareza, heterogeneidad genética y asociaciones con síndromes de insuficiencia de la médula ósea. Los avances recientes en la comprensión genética, la clasificación y las estrategias de tratamiento han mejorado los resultados y allanado el camino para enfoques personalizados. Si bien el TCMH alogénico sigue siendo el tratamiento de elección para muchos casos, la terapia inmunosupresora proporciona una alternativa para pacientes seleccionados con CCR.
La investigación y la colaboración continuas son esenciales para refinar los criterios de diagnóstico, mejorar la eficacia del tratamiento y, en última instancia, ofrecer una mejor calidad de vida para los niños afectados por estos trastornos desafiantes.